Dra. María Edilma Araúz Araúz. Departamento de Anatomía Patológica.
Dr. Michael Pimentel. Dra. Graciela Tejedor. Servicio de Pediatría
Hosp. Materno Infantil José Domingo De Obaldía. Chiriquí – Panamá
Paciente masculino de 13 años de edad, con historia de 1 año de evolución, caracterizada por pérdida de peso (aproximadamente 10 kg), astenia y adinamia. En los 30 días previos a su hospitalización presento masa palpable en abdomen, tos seca, anorexia, vomito (#1) y diarrea sin sangre, ni moco (#1).
Antecedentes patológicos personales negados. Antecedente familiar: diabetes mellitus (madre).
Niega hospitalizaciones previas.
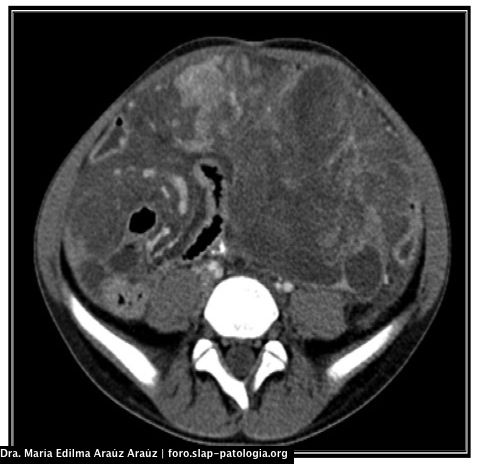
Examen físico: afebril, alerta. Peso: 40Kg, talla: 155 cm, presión arterial: 100/60 mmHg, frecuencia cardíaca: 95 x’, frecuencia respiratoria: 16 x’; saturación de oxigeno: 100%. Como hallazgo, en abdomen: se palpa masa en hipocondrio izquierdo hasta área umbilical de consistencia dura, dolorosa a la palpación profunda y adherida a planos profundos, de más o menos 13 cm x 7 cm, no megalias.
Laboratorios:
Biometría hemática: hemoglobina: 12.2g/dL, leucocitos: 14,800, neutrófilos: 73.6%, linfócitos: 16.6%, plaquetas: 401,000. TP: p 18.3 C 16.0; TPT: p: 27.5 C: 32.5; fibrinógeno: p 657 c 344.
Urinálisis normal
Química sanguínea:
| Proteinas: 7.48 g/dL |
| Albumina: 3.87 g/ dL |
| Acido Urico: 2.82 mg/dL |
| Bilirrubina Total: 1.11 mg/dL |
| AST: 16.4 U/L |
| ALKP: 118.7 U/L |
| ALT: 11.0 U/L |
| LDH: 667.5 |
| Bil. Conjugada 0.0 mg/dL |
| Ig M: 127.73 mg/dL |
| Ig A: 160.24 mg/dL |
| Globulina: 3.6 g/dL |
| Ferritina: ho hay reactivo |
| Frotis de sangre periférica: normal |
| Na: 136 mmol/L |
| K: 4.4 mmol/L |
| Ca: 9.3 mg/dL |
| Cl: 95 mmol/L |
| Glucosa: 97 mg/dL |
| BUN: 7 mg/dL |
| Creatinina: 0.6 mg/dL |
| Amilasa: 34 U |
| AL KP: 151 U/L |
| PROT –C : 9 mg/dl |
VDRL: negativo; VIH: negativo AFP: 3.70ng/dL; CA19-9 XR: < 2.00, CA125: 213.7 U/ml
Se toma biopsia percutánea (imagenes proporcionadas).



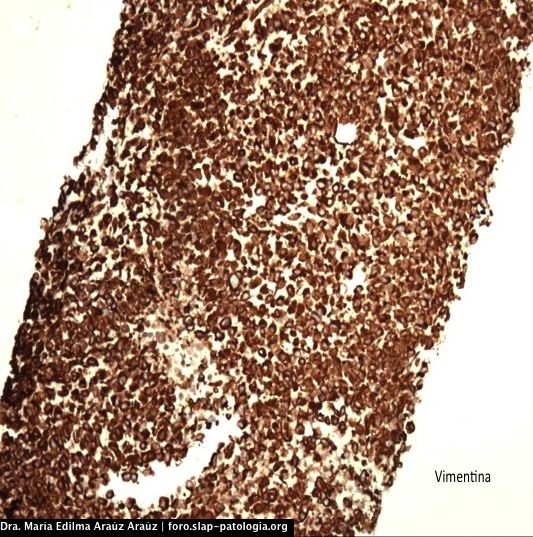
DIAGNÓSTICO:
CONCLUSIÓN DEL CASO:
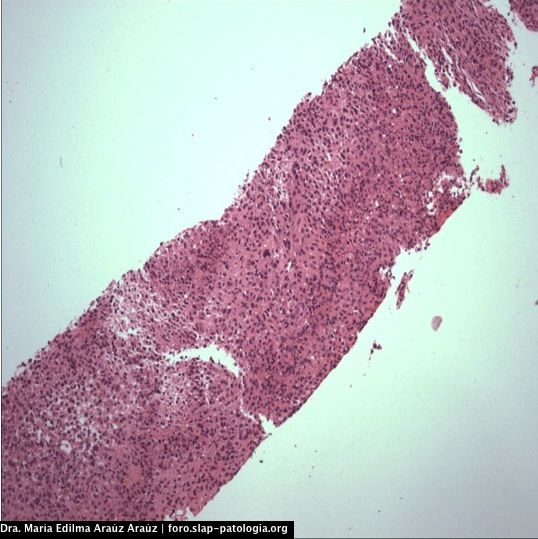
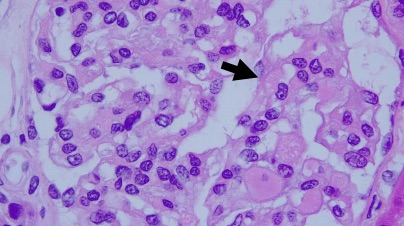
REGION DE ABDOMEN, TEJIDOS BLANDOS (BIOPSIA PERCUTANEA):
NEOPLASIA FUSOCELULAR MALIGNA.
Vimentina positivo. S-100 negativo. CKAE1/AE3: negativo, CD45: negativo.
POR LAS CARACTERISTICAS HISTOLOGICAS Y SU ASOCIACION CON NEUROFIBROMATOSIS TIPO 1, LA NEOPLASIA ES CONSISTENTE CON TUMOR DE LA VAINA NERVIOSA PERIFERICA MALIGNO.
DISCUSION:
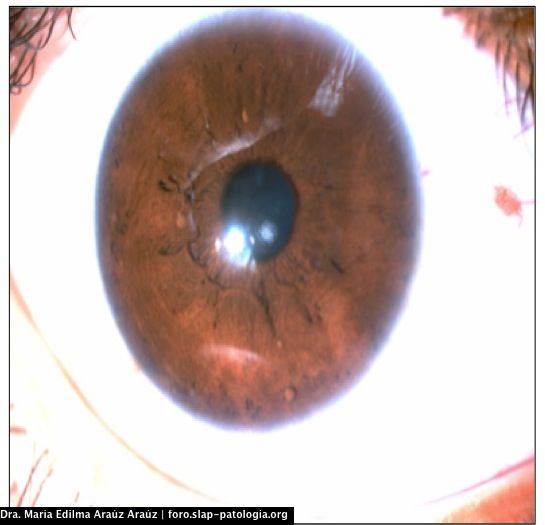
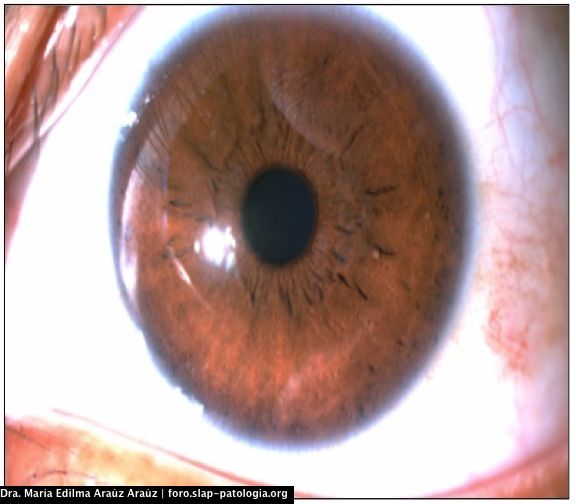
El paciente cumple con criterios diagnósticos para neurofibromatosis tipo 1: en la piel presentaba manchas café con leche en extremidades, tronco, axilas y áreas inguinales mayores de 1.5 cm y efélides en área inguinal y axilar. La evaluación oftalmológica reportó nódulos de Lisch en ambos ojos.
El paciente fue enviado a hospital de tercer nivel, donde recibió quimioterapia y manejo oncológico por 5 meses, durante los cuales se realizó nueva biopsia la cual se reporta:
- Neurofibroma con cambios malignos focales ( Tumor de vaina nervioso Periférico Maligno)
En laparotomía posterior se observa extensión tumoral hacia intestino, no resecable. Asoció lesiones de apariencia metastásica en hígado. Se deriva a manejo paliativo, fallece dos meses después.
La Neurofibromatosis tipo 1 (Enfermedad de von Recklinghausen o NF1) es causada por la mutación del gen NF1 en el cromosoma 17q11.2. Se hereda con patrón autosómico dominante. El 50% de los pacientes diagnosticados presenta mutaciones espontáneas (no familiar afectado, como en nuestro caso). El gen NF1 codifica para la proteína neurofibromina, regulador importante en la vía de proliferación celular de RAS que se expresa en todo el organismo, explicando las manifestaciones sistémicas de la enfermedad; la inactivación del gen NF1 se implica en la proliferación tumoral en estos pacientes.
El diagnóstico de NF1 es clínico para la mayoría de los pacientes, las pruebas genéticas son reservadas para casos dudosos o en contexto de estudios de investigación. El National Institutes of Health (NIH) estipula que el paciente tenga al menos 2 de los criterios diagnósticos establecidos.
Table 1: National Institutes of Health (NIH) diagnostic criteria for neurofibromatosis type 1 (NF1)
Six or more café-au-lait macules >5mm in greatest diameter in prepubertal individuals, and >15mm in postpubertal individuals
Two or more neurofibromas of any type or one plexiform neurofibroma
Freckling in the axillary or inguinal regions
Optic glioma
Two or more iris hamartoma (Lisch nodules)
Distinctive bony lesion such as sphenoid dysplasia, or thinning of the long bone cortex with or without pseudoarthrosis
A first-degree relative (parent, sibling or offspring) with NF1 based on the above criteria
La inactivación del gen NF1 (por ejemplo, por mutación) estaría implicada en la proliferación tumoral en estos pacientes. El tumor de la vaina nerviosa periférica maligno, previamente llamado neurofibrosarcoma, es la principal causa de morbilidad y mortalidad en NF1, se origina típicamente de transformación maligna de neurofibromas plexiformes y ocasionalmente de las raíces de nervios espinales o neurofibromas subcutáneos. El riesgo de desarrollarlo en NF1 es de 8-13%. Otros tumores asociados a NF1 son: gliomas de nervio óptico, rabdomiosarcomas, neuroblastomas y leucemia mielomonocítica juvenil en pacientes pediátricos; así como tumor del estroma gastrointestinal, feocromocitomas, gliomas de nervio óptico y tumores carcinoides en adultos. Se ha asociado a tumores de mama, pulmón, ovario, cerebro, melanoma, carcinoma colorrectal, entre otros. Sea reportado prevalencia del TVNPM hasta de 43%.
La histología del tumor de la vaina nerviosa periférico es constituida por células fusiformes en forma de coma con arreglo de fascículos hipocelulares y áreas mixoides entrecruzadas, nódulos o remolinos de células fusiformes, palizada en 10% de casos, asocia áreas geográficas de necrosis con palizada en los bordes, áreas hemangiopericitoides. La inmunohistoquímica es positiva para S-100 hasta en 68% de casos (nuestro caso fue negativo, lo cual podría estar relacionado con el pequeño tamaño del especímen al que se le pudo realizar inmunohistoquímica), es positivo para vimentina (como en nuestro caso). Se establece que una fuerte y difusa reactividad para S-100 en un tumor de células fusiformes maligno sugestivo de TVNPM debe elevar la sospecha de diagnóstico alternativo de melanoma, particularmente cuando la lesión se ubica en piel o ganglio linfático. Se describe que algunas de estas neoplasias pueden tener diferenciación divergente rabdomioblástica (tumor de Tritón maligno), osteosarcoma, condrosarcoma, angiosarcoma.
Hay dos circunstancias en las cuales se debe tener al tumor de la vaina nerviosa periférica maligno como consideración primaria de un tumor maligno de tejidos blandos constituido por células fusiformes:
1- Cuando el tumor se desarrolla en un paciente con enfermedad de von Recklinhausen tipo 1 (como en nuestro paciente)
2- Cuando el tumor tiene obvio origen en compartimiento anatómico de un nervio principal o en continuidad con un neurofibroma.
La sobrevida a los 5 años es de 15 % a 30 %, nuestro paciente falleció a los 7 meses de su diagnóstico. El tratamiento de elección es la resección completa con márgenes amplios, que depende principalmente de la localización.
Bibliografía:
1- Yoon-Sim Yap, John R. McPherson, Choon-Kiat Ong, Steven G. Rozen, Bin- Tean Teh, Ann S. G. Leeand David F. Callen The NF1gene revisited-from bench to beside. Oncotarget, Advance Publications 2014. www.impactjournals.com/oncotarget/
2- Man-Kyu Park, M.D., Joo-Kyung Sung, M.D., Ph.D., Kyung-Hun Nam, M.D., Kyoung-Tae Kim, M.D. Malignant Peripheral Nerve Sheath Tumor of Non-Neurofibromatosis Type I Metastasized to the Cerebrospinal Axis. J Korean Neurosurg Soc 53 : 190-193, 2013.
3- Baena-Ocampo L, Reyez-Sanchez A, Alpizar-Aguirre A, Rosales-Olivares L. Tumor de la vaina nerviosa periférica maligno asociado a neurofibromatosis tipo 1. Informe de 2 casos. Cir Ciruj 2009.77:391-395
4- Rosai and Ackerman´s Surgical Pathology. Volume 2. Tenth edition. Elsevier Mosby. China. 2011.

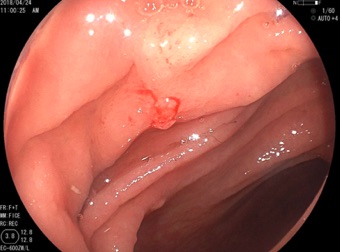
El niño porta una NF tipo 1, ya que se muestran máculas café con leche, lesiones axilares, nódulos de Lisch.
La NF es un sindrome de predisposición tumoral y, entre las neoplasias más frecuentes a las que predispone, se hallan los tumores de la vaina de los nervios periféricos, sarcomas, rabdomiosarcomas y tumores del estroma intestinal, los cuales son los tumores mesenquimales más frecuentes del tracto gastrointestinal. Y algo así es la masa…una proliferación fusocelular….
Dra. Di Martino, la correlación clínica es muy importante, algo que no debemos olvidar. Gracias por su comentario
Coincido con Beatriz Di Martino. Por morfología y frecuencia considero que se pueda tratar de un tumor maligno de la vaina del nervio periférico. Definitivamente es un sarcoma por lo que la expresión de vimentina se explica. Es necesario ampliar el panel.
Tumos fuso celular , lo mas probable y con base en la historia clínica tumor maligno de la vaina de nervio periferico. Amplio el estudio con estudios de IHQ incluido el ki67