
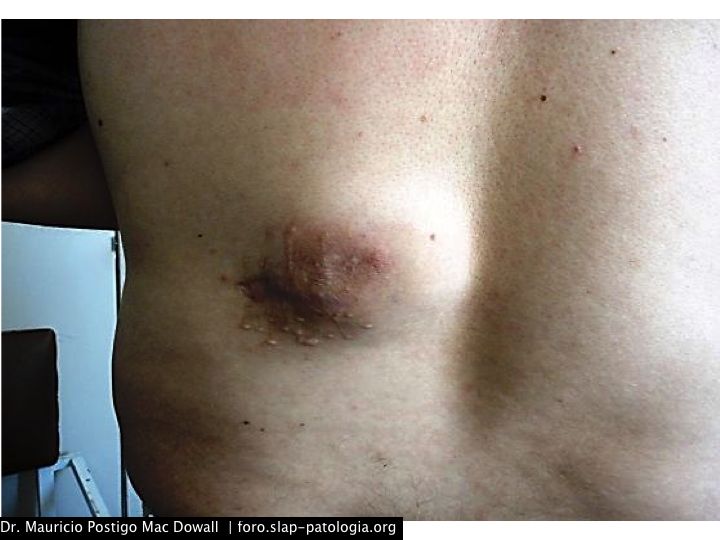
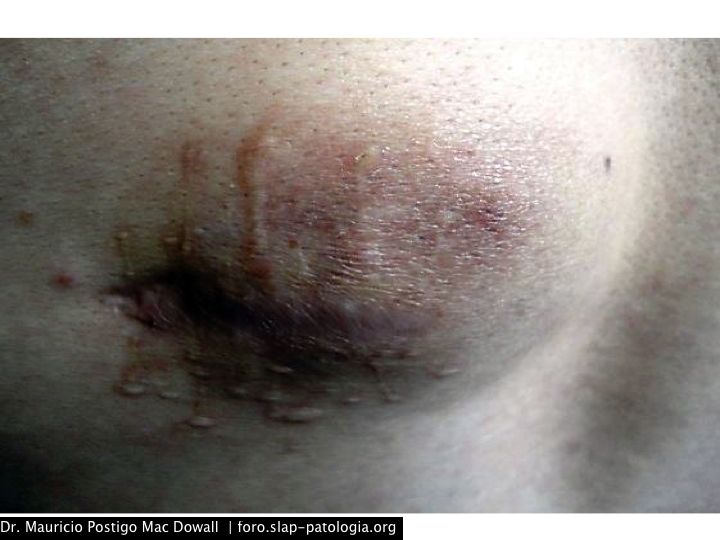
Paciente varón de 33 años, natural y procedente de Arequipa-Perú. Ingeniero químico de profesión. Inició su enfermedad en el 2008, con una lesión máculo-papular eritematosa en la piel de la región lumbar izquierda, al poco tiempo de iniciar su trabajo en una empresa de materiales de escritorio, donde estaba en contacto con pinturas, solventes y materiales relacionados. Se realizó una resección incompleta. Luego de 6 a 8 meses notó un aumento de la masa, tornándose una placa y luego nódulo eritematoso con compromiso del tejido adiposo subcutáneo. Se le realizó una resección completa de la lesión, y luego de un año ésta volvió a crecer en forma de nódulo eritematoso de 12 cm de diámetro mayor. Desde hace cuatro meses nota la presencia de dos masas adicionales en el tejido adiposo subcutáneo de la cara ántero-inferior del tórax y en el abdomen, de 0,9 y 1 cm, respectivamente.
- En la analítica sanguínea no muestra alteraciones significativas del conteo hematológico ni alteraciones bioquímicas. Pruebas virales (HIV, HTLV, EBV) negativas.
- Mediante ecografía se apreciaba lesión en partes blandas del dorso que mide 32 x 22 mm con bordes definidos y flujo periférico con algunas áreas fibróticas. Otra lesión que mide 9 x 6 mm en la pared abdominal anterior de mayor ecogenicidad en el plano superficial.
- Tomografía de tórax, abdomen y pelvis dentro de rangos normales. No se evidenciaban adenopatías. TEM cerebral normal. Mediante una TEM de abdomen superior se apreció una tumoración en el tejido adiposo subcutáneo dorso-lumbar izquierdo a nivel de D12 de 38 x 13 x 25 mm, de 28 UH sin contraste y 59UH con contraste. No se evidenciaban adenopatías ni lesiones adicionales.
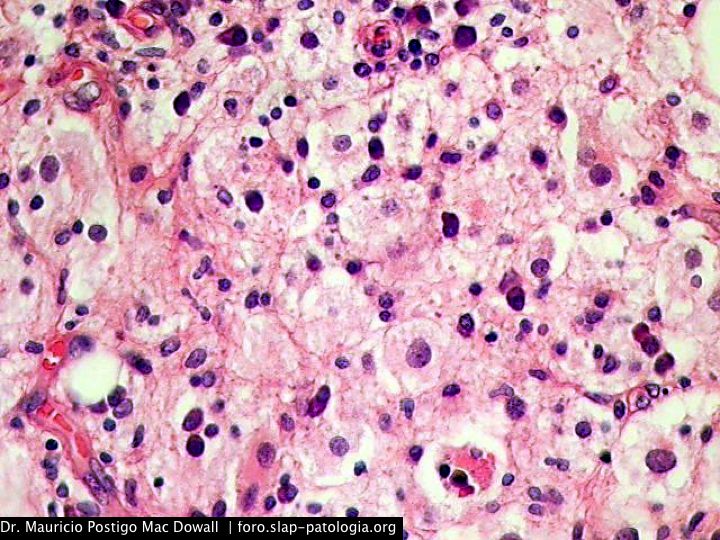
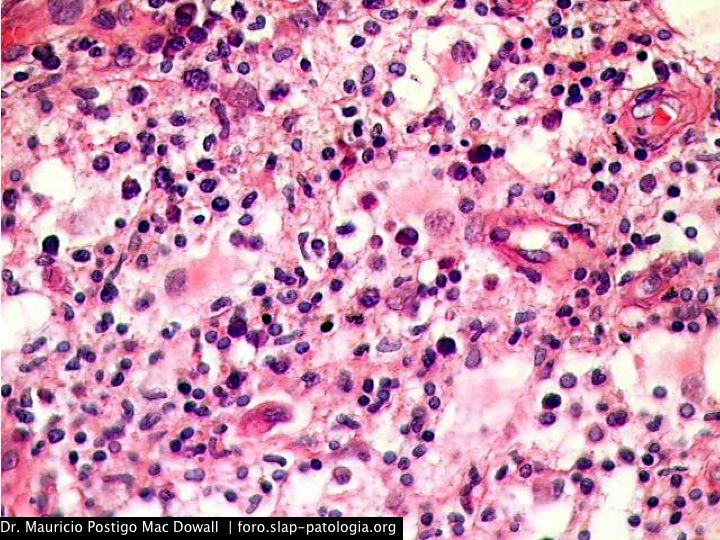
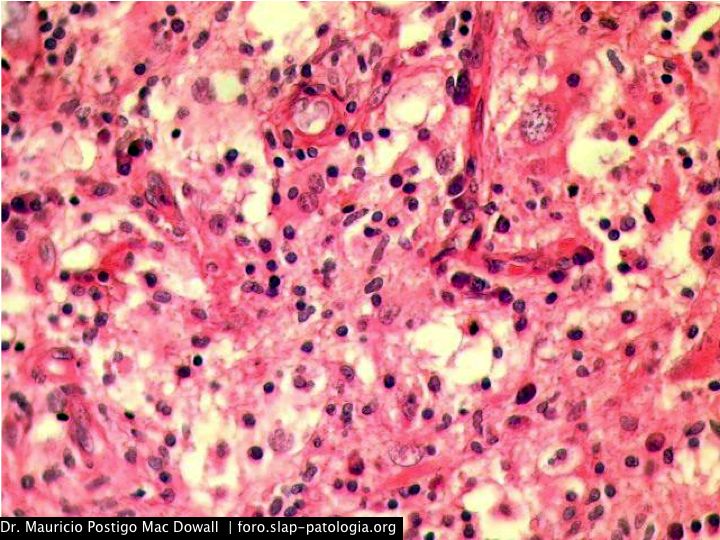
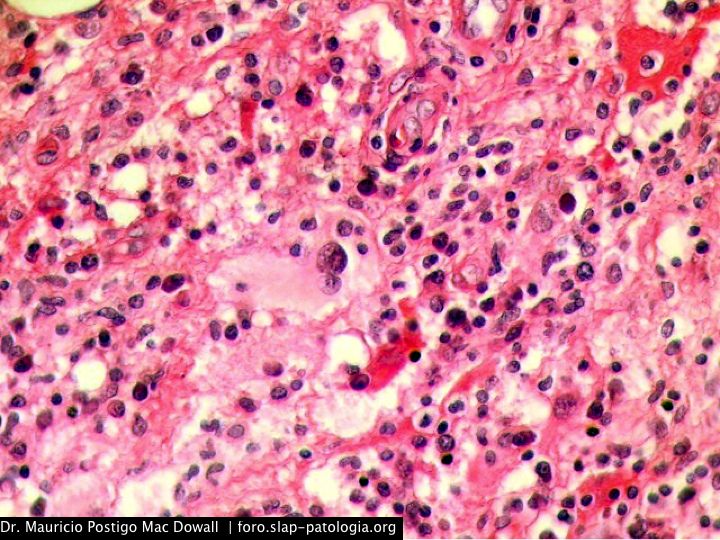
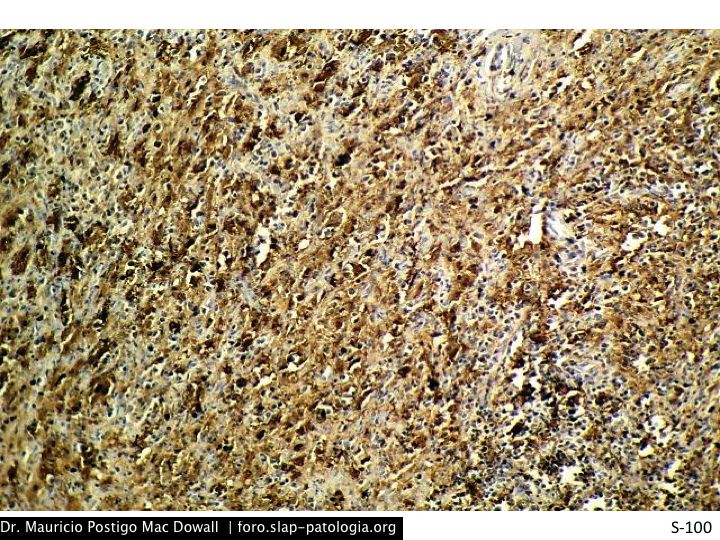
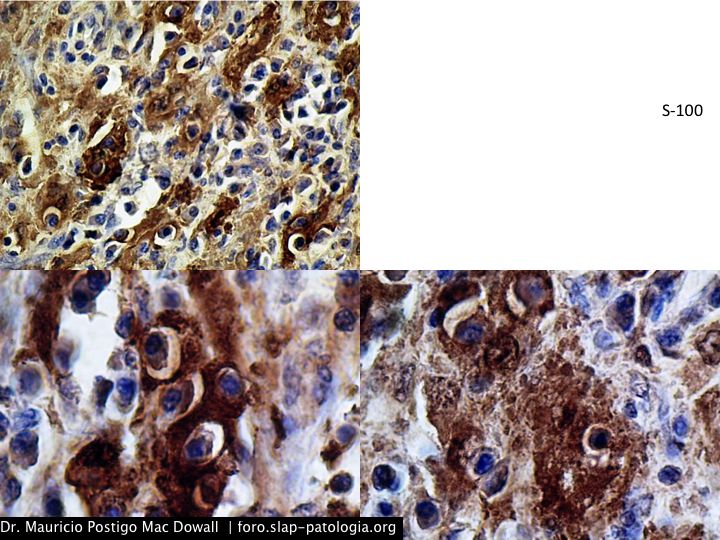
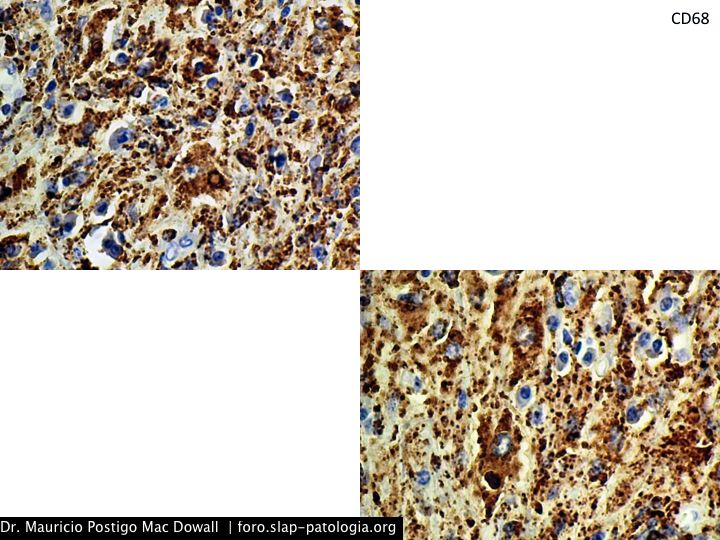
TINCIONES DE INMUNOHISTOQUIMICA
CUAL ES SU DIAGNOSTICO?
DIAGNÓSTICO:
ENFERMEDAD DE ROSAI DORFMAN
DISCUSIÓN
La enfermedad de Rosai Dorfman en una enfermedad no neoplásica rara. La forma cutánea y extracutánea es mayormente vista en personas entre la tercera y quinta décadas, mujeres (2;1 en relación a los hombres), siendo la edad media de 43.5 años, y afecta a gente de raza asiática y blanca3,4. Los ganglios linfáticos/enfermedad sistémica son predominantemente afectados niños y adultos jóvenes varones (1.4:1 en relación a mujeres), siendo la edad media de 20.6 años, y raramente son de raza asiática. La etiología es desconocida. Las lesiones son policlonales, probablemente secundarias a una disrregulación inmune.
Se ha descrito una asociación a HHV-6, encontrándose DNA específico del HHV-6 en los histiocitos de pacientes con ERD mediante hibridización in situ en algunos casos, así como la expresión de la proteína ORF1 en los histiocitos mediante inmunohistoquímica.5,11 El Parvovirus B19 también ha sido implicado, encontrándose expresión de las proteínas de la cápside VP1/VP2 mediante inmunohistoquímica.6
Lo más característico es el compromiso de los ganglios linfáticos cervicales. Los niños con adenomegalias cervicales masivas frecuentemente sufren de fiebre y malestar general. Los exámenes de laboratorio muestran leucocitosis, anemia, hipergamaglobulinemia policlonal e incremento de la VSG.8
En el 43% de los casos, se observa evidencia de compromiso extranodal simultáneamente, y en sólo el 23% ocurre ERD extranodal aislada. Los sitios de compromiso extranodal en orden decreciente son: piel y tejidos blandos (16%); cavidad nasal y senos paranasales (16%); ojos, órbita y anexos oculares (11%); hueso (11%); glándula salival (7%); sistema nervioso central (7%); cavidad oral (4%); riñón y tracto génito-urinario(3%); tracto respiratorio (3%); hígado (1%); amígdala (1%); mama (<1%); tracto gastrointestinal (<1%); y corazón (<1%).9,12
Las formas cutáneas puras son raras, tomando la forma de pápulas solitarias, agrupadas o ampliamente difundidas; raramente se pueden ver placas y nódulos. La regresión de las lesiones dejan máculas atróficas y marrones.7 Las lesiones cutáneas ocurren más frecuentemente en la cabeza y el cuello; las lesiones mucosas en la nariz y en los senos paranasales.
En el estudio más grande de ERD, el 3% presentaba la típica morfología de la ERD del tejido blando sin linfadenopatía detectable.12 Un estudio retrospectivo conducido por el Instituto de Patología de las Fuerzas Armadas de Estados Unidos describió 17 casos de ERD del tejido blando, 13 de los cuales no presentaron linfadenopatías.13 Las formas en el tejido blando se presentan como tumoraciones subcutáneas firmes no dolorosas cuyo tamaño varía de 1 a 10 cm, sobre cualquier área del cuerpo.4
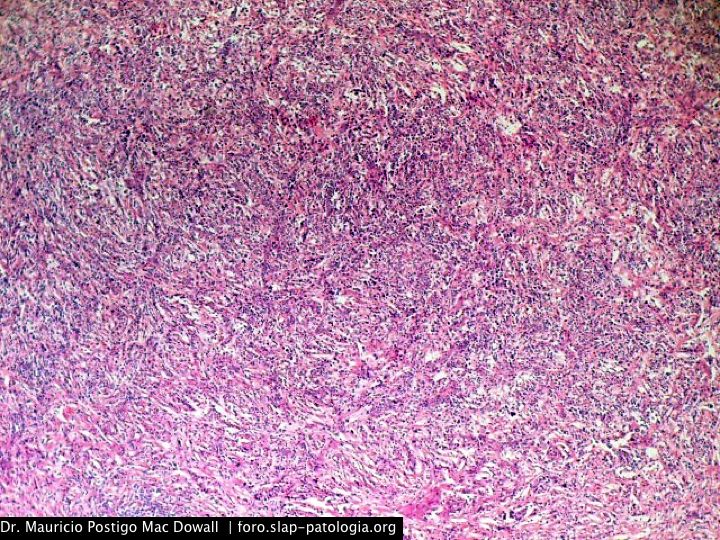
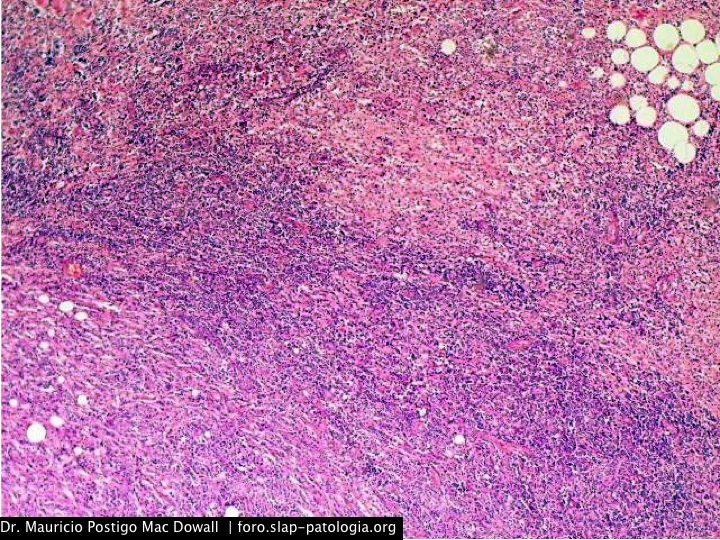
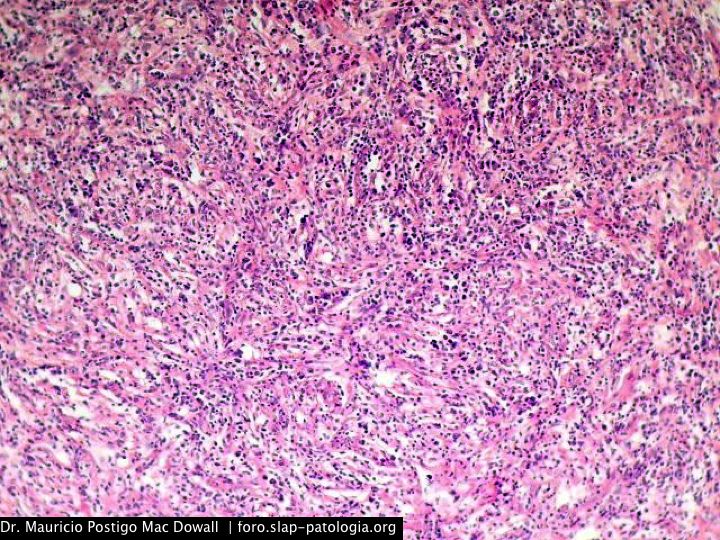
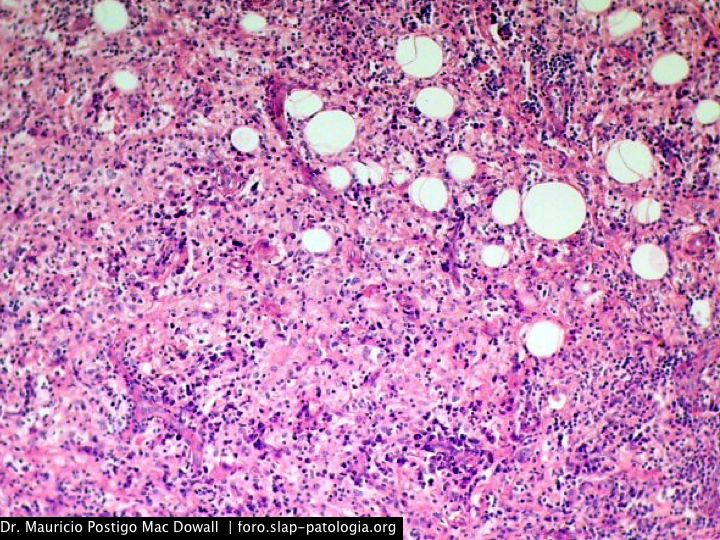
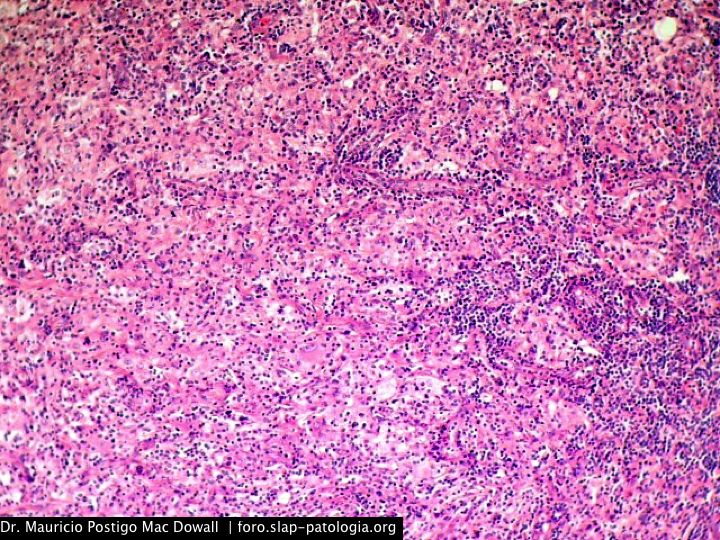
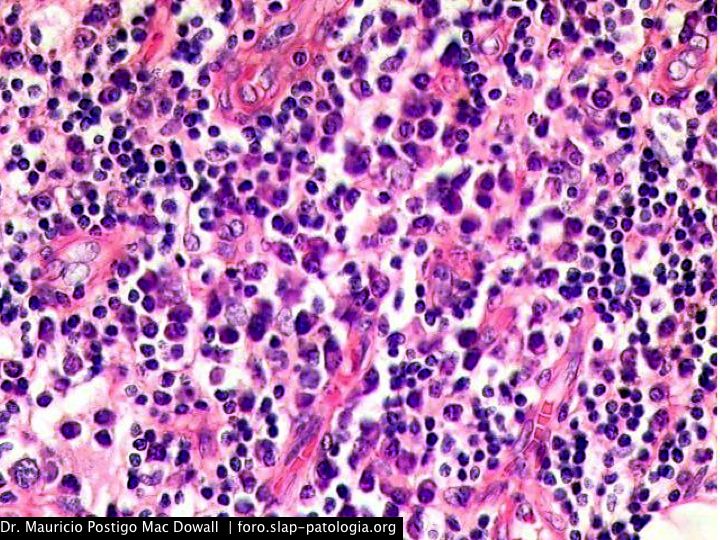
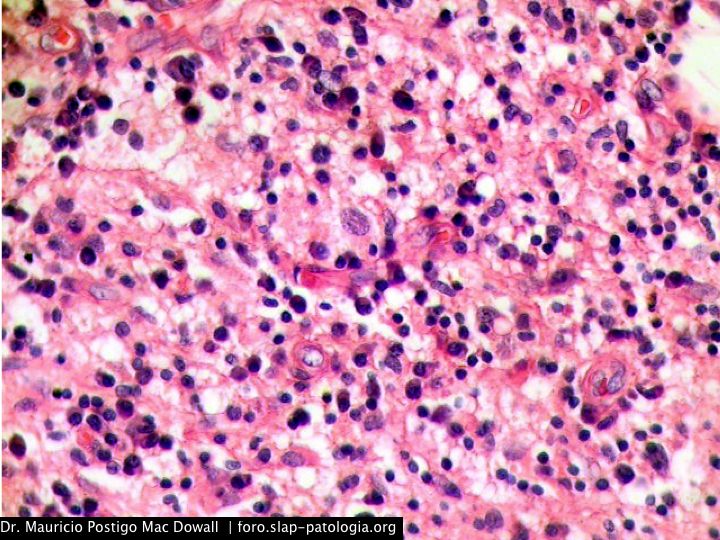
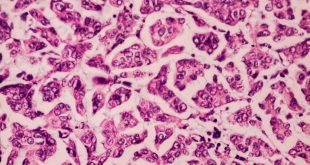
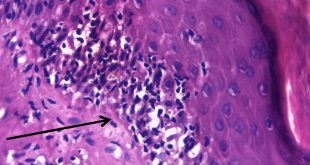
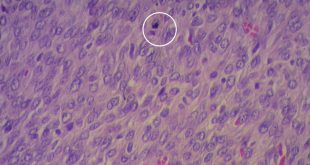
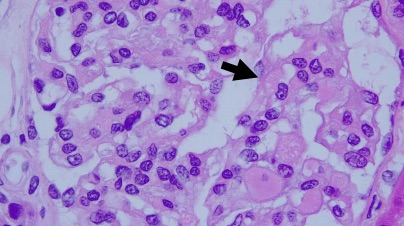
En el estudio histopatológico de los ganglios linfáticos, la arquitectura es reemplazada por mantos de macrófagos débilmente teñidos a ligeramente eosinofílicos a nivel de la zona sinusal. En las localizaciones extranodales los infiltrados simulan a los senos de los ganglios linfáticos (patrón sinusoidal). Se evidencia una prominente emperipolesis de linfocitos, eritrocitos u otras células, pero no es específica; puede ser vista también en otras condiciones, como en linfomas de células T subcutáneo. Se pueden encontrar linfocitos, células plasmáticas, neutrófilos y fibroesclerosis en varios grados. En las pruebas de inmunohistoquímica los macrófagos son positivos para CD68 (PGM1, KP1) y S100; CD1a, factor XIIIa y CD34 son negativos. En microscopía electrónica los macrófagos ingieren linfocitos intactos. Se evidencian las estructuras fagolisosómicas, mas no los gránulos de Birbeck.7
Morfológicamente, la ERD extranodal, especialmente en su forma cutánea, debe incluir en su diagnóstico diferencial al xantoma eruptivo, enfermedad de Tangier, histiocitosis de células de Langerhans, retículohistiocitoma, xantogranuloma juvenil, lepra lepromatosa, linfoma de Hodgkin, y no Hodgkin, histiocitosis maligna y un pseudolinfoma.1 Las características morfológicas e inmunohistoquímicas distintivas de la ERD, las tinciones realizadas para cadenas ligeras kappa y lambda y para CD30 descartan las posibilidades diagnósticas diferenciales en nuestro caso.
En cuanto al pronóstico, la manifestación en niños y el compromiso de ganglios linfáticos están asociados a regresión más probable y rápida, en comparación a la presentación en adultos y la extensión a sitios extranodales. La gran mayoría de las lesiones son autolimitadas y benignas. Las raras fatalidades han sido asociadas a desórdenes inmunológicos, linfomas de Hodgkin y no Hodgkin, leucemias, y casos excepcionales con tumores sólidos.7
En cuanto al tratamiento, múltiples modalidades han sido utilizadas, incluyendo a la escición quirúrgica, glucocorticoides sistémicos, talidomida, crioterapia, radioterapia, interferón alfa y aciclovir, en general con buenos resultados.
BIBLIOGRAFÍA
1. Huang HY, Yang CL, Chen WJ. Rosai-Dorfman Disease with Primary Cutaneous Manifestations—A Case Report. Ann Acad Med Singapore 1998;27:589-93.
2. Chopra D, Svensson WE, Forouhi P, y Poole S. A rare case of extranodal Rosai-Dorfman disease. Bri J Radiol 2006;79:117–119.
3. Zannolli R, Acquaviva A, Polito E, Galluzzi P, y col. Pathological Case of the Month. Multifocal Rosai-Dorfman Disease of Soft Tissue. Arch Pediatr Adolesc Med 1999;153:1199-1200.
4. Penna Costa AL, Oliveira e Silva N, Pamponet Motta M y col. Soft tissue Rosai-Dorfman disease of the posterior mediastinum. J Bras Pneumol. 2009;35:717-720.
5. Levine PH, Jahan N, Murari P, et al. Detection of human herpesvirus 6 in tissues involved by sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). J Infect Dis 1992;166:291–5.
6. Mehraein Y, Wagner M, Remberger K y col. Parvovirus B19 detected in Rosai–Dorfman disease in nodal and extranodal manifestations. J Clin Pathol 2006;59:1320–1326.
7. Zelger B, Kohler S, Burgdorf W. Chapter 4. Haematolymphoid tumours. World Health Organization Classification of Tumours: Pathology and Genetics of Skin Tumours. IARC press, Lyon 2006; p221.
8. Yilmaz S, Ture M, Maden A y Tunakan M. Extranodal Rosai-Dorfman disease with bilateral orbital involvement: Report of a case treated with systemic steroid alone. Clin Ophthal 2008:2(2) 479–481.
9. Shi S, Sun Y y Guo L. Rosai-Dorfman disease of lung: a case report and review of the literatures. Chinese Med J 2009;122(7):873-874.
10. Miyake M, Tateishi U, Maeda T, Arai Y, Sugimura K, Hasegawa T. Extranodal Rosai-Dorfman disease: a solitary lesion with soft tissue reaction. Radiat Med 2005; 23:439-442.
11. Luppi M, Barozzi P, Garber R, Maiorana A, Bonacorsi G, ArtusiT, y col. Expression of human herpesvirus-6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol 1998; 153: 815-823.
12. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7(1):19-73.
13. Montgomery EA, Meis JM, Frizzera G. Rosai-Dorfman disease of soft tissue. Am J Surg Pathol. 1992;16(2):122-9.

CD34 se le hizo?
No se le realizó ese marcador.
Se trata de un tumor de celulas de Langerhans (histiocitosis X)
Enfermedad de Rosai-Dorfman. (Una clave presencia de emperipolesis )
Y si che, una Enfermedad de Rosai Dorfman cutánea es, no asociada a HTLV por lo visto…rara. Lo de la emperipolesis aparece pero no es patognomónico. Se ve sobre todo en la paniculitis histiocitica citofagica sistemica, Síndrome de Sweet histiocitoide, entre otras…