
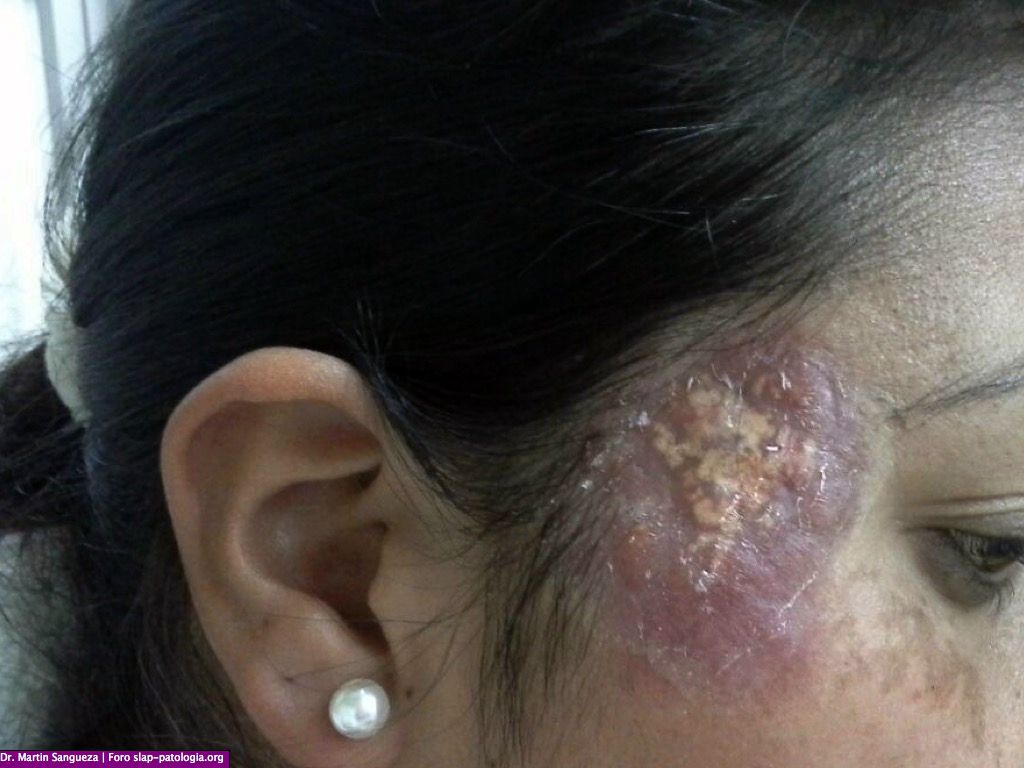
Paciente de sexo femenino 32 a. con una lesion tumoral en region temporo-malar derehca. La lesion es progresiva y tiene 1 ano de evolucion.



¿Cuál es tu diagnóstico?
DIAGNÓSTICO:
Enfermedad de Rosai Dorman
Discusion
Clásicamente la enfermedad de Rosai-Dorfman tiene dos formas: la sistémica, también denominada histiocitosis sinusal con linfadenopatía masiva o enfermedad de Rosai- Dorfman sistémica y la extra nodal cuyo principal órgano blanco es la piel, además de tracto respiratorio superior, entre otros1-4.
La enfermedad de Rosai-Dorfman cutánea es muy rara, representa el 3% del total de casos, afecta adultos mayores de 40 años, predominando en mujeres. Su etiología es desconocida pero se postula una posible causa infecciosa, en la cual estarían implicados el VEB y el VHS-6 y VHS-8. Aparentemente el cuadro clínico sería un fenómeno de hiperrespuesta anormal de los histiocitos a estos virus, sin embargo para otros autores estas infecciones virales serían secundarias al desorden primario del sistema inmune. Las pruebas serológicas positivas para estos virus no son concluyentes acerca de su probable rol etiológico, por lo que se recomienda la hibridización in situ para su detección en los tejidos.
El cuadro clínico de la forma cutánea se caracteriza por su evolución crónica, con remisiones y exacerbaciones, o por ser indolora y autolimitada. En los reportes los pacientes permanecen asintomáticos y en buen estado general. En el compromiso de piel existe polimorfismo clínico, pueden presentarse máculas, pápulas, nódulos o placas infiltradas eritematosas o violáceas, xantomatosas o pardo-amarillentas, aisladas o diseminadas. Puede aparecer en cualquier área anatómica, sin embargo las zonas más frecuentemente comprometidas son los parpados y la región malar. A menudo se reporta anemia moderada, velocidad de sedimentación eritrocitaria elevada e hiperganmaglobulinemia policlonal en estos pacientes, lo cual soporta la teoría de una respuesta inmunológica exagerada a agentes infecciosos.
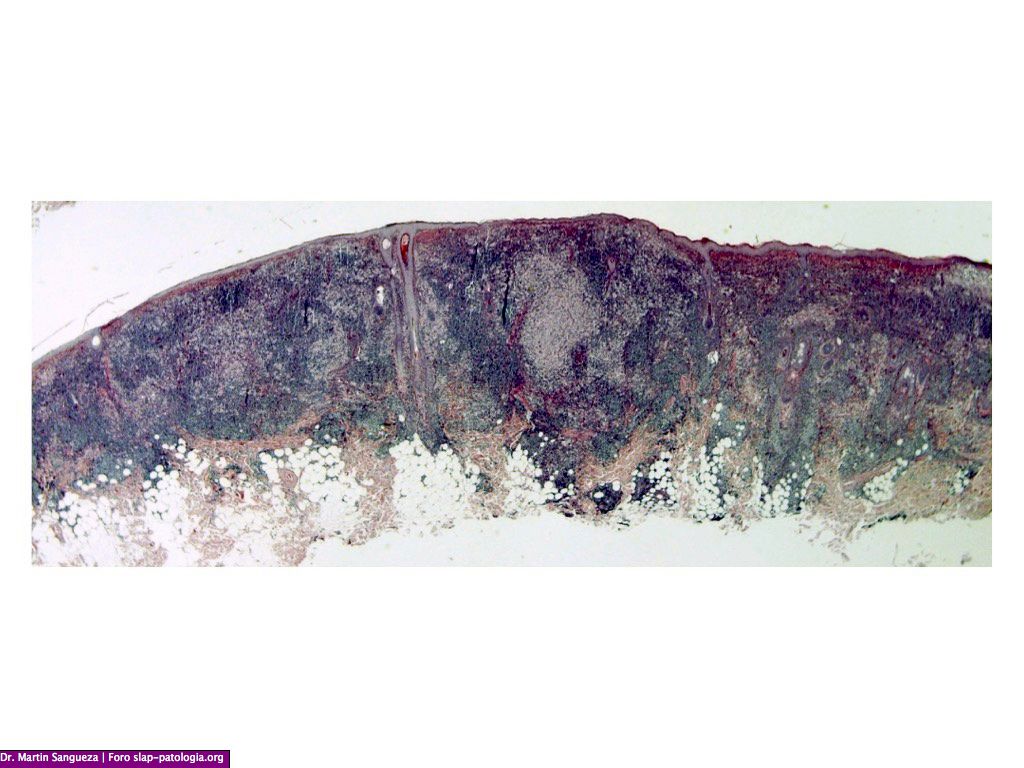
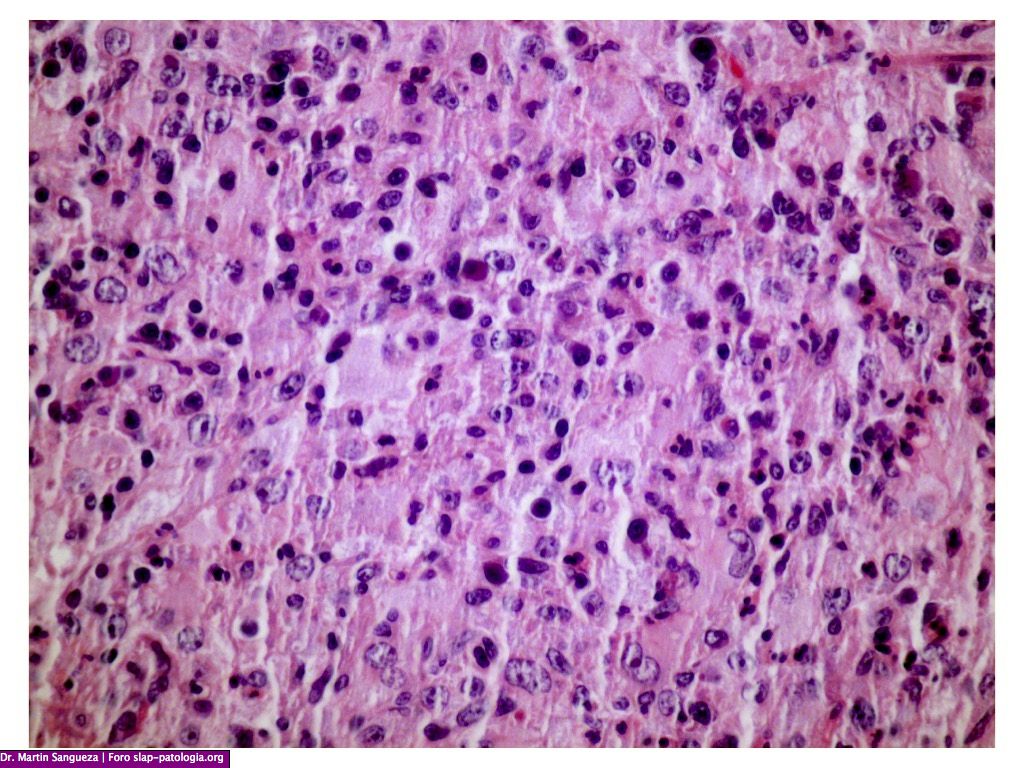
El diagnóstico clínico es difícil, por lo que se requiere del examen histológico y de inmunohistoquímica; en la biopsia observamos una epidermis normal y en dermis un infiltrado denso de histiocitos grandes, citoplasma eosinofílico amplio, con un núcleo grande y vesicular, además de linfocitos y células plasmáticas, destacando el fenómeno de emperipolesis o fagocitosis de células inflamatorias intactas en el citoplasma de los histiocitos, algunas dentro de vacuolas fagocitica. Estos hallazgos son menos frecuentes en la forma cutánea que en la sistémica dificultando aún más el diagnóstico, La inmunohistoquímica muestra que estos histiocitos son S100 y CD68 positivos pero CD1a negativos. El diagnóstico diferencial se debe de realizar con los siguientes cuadros: histiocitosis, sarcoidosis, infecciones o procesos infiltrativos tumorales, en nuestra paciente se plantearon además de estos diagnósticos los de policondritis recidivante y pseudolinfoma.
El tratamiento depende de la evolución, pues hay casos de resolución espontánea, no se ha reportado que la forma cutánea progrese a un compromiso sistémico. Los tratamientos reportados incluyen corticoides tópicos, intralesionales y sistémicos como prednisolona, metilprednisolona y dexametasona, metotrexate, mercaptopurina.
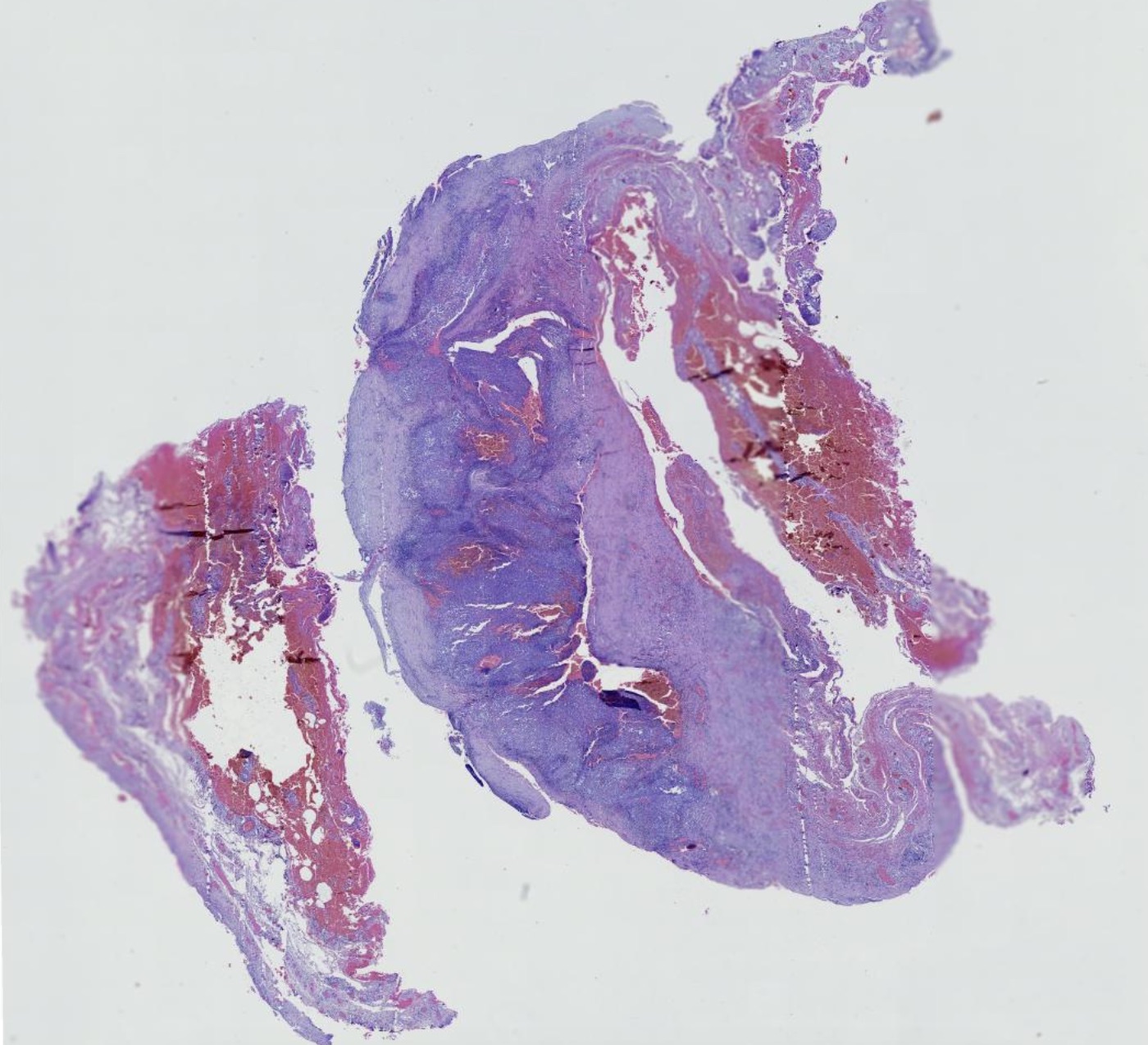
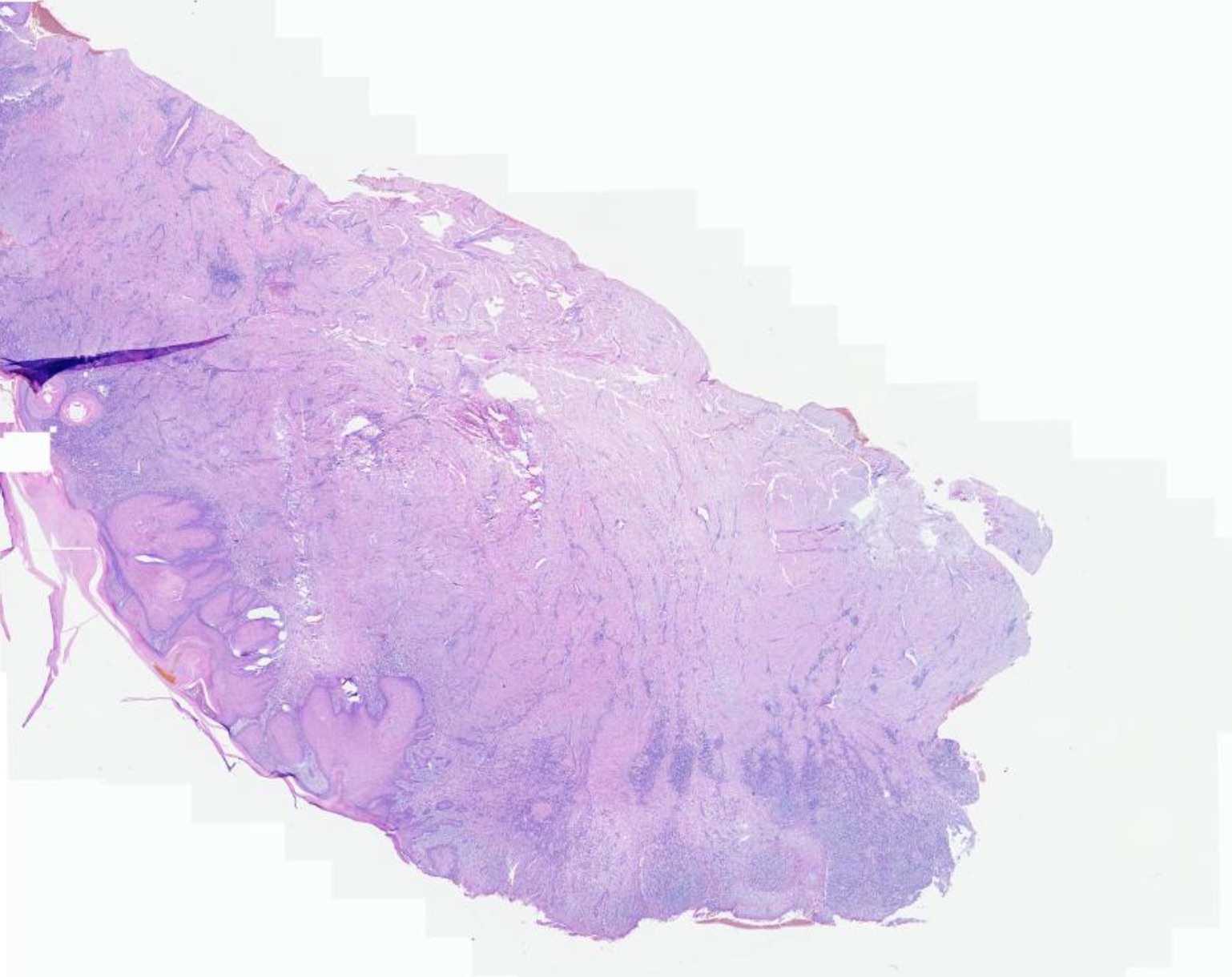

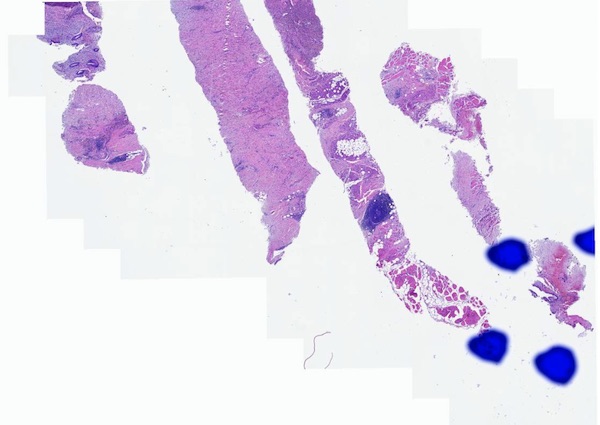
Rosai-Dorfman
Histiocitosis de células de langerhans