
M. Adelita Vizcaíno M.D.
Profesora Titular
Departamento de Biología Celular y Tisular, Facultad de Medicina, Universidad Nacional Autónoma de México, D.F., México
Fausto J. Rodríguez M.D.
Profesor Asociado
División de Neuropatología, Johns Hopkins University, USA
Presentación Clínica
Hombre de 44 años.
Padecimiento actual de 18 meses con diplopía, nistagmus y ataxia progresivos.
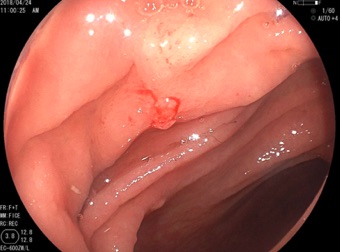
La resonancia magnética demostró una lesión hipercaptante en la fosa posterior, que fue extirpada.

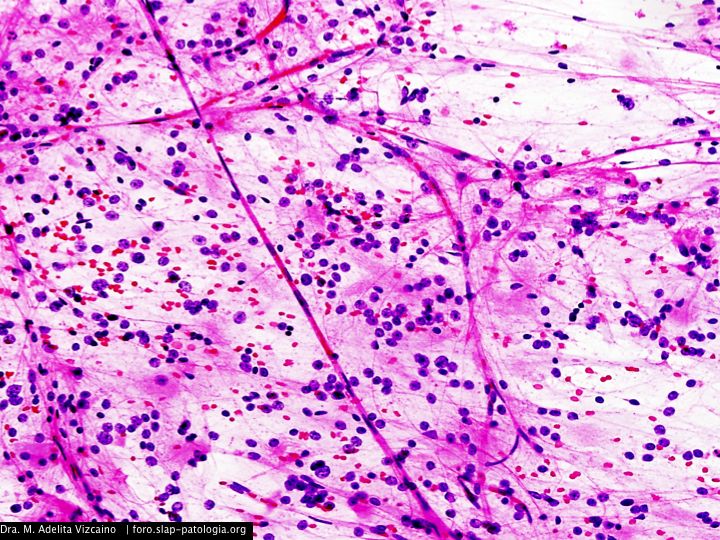


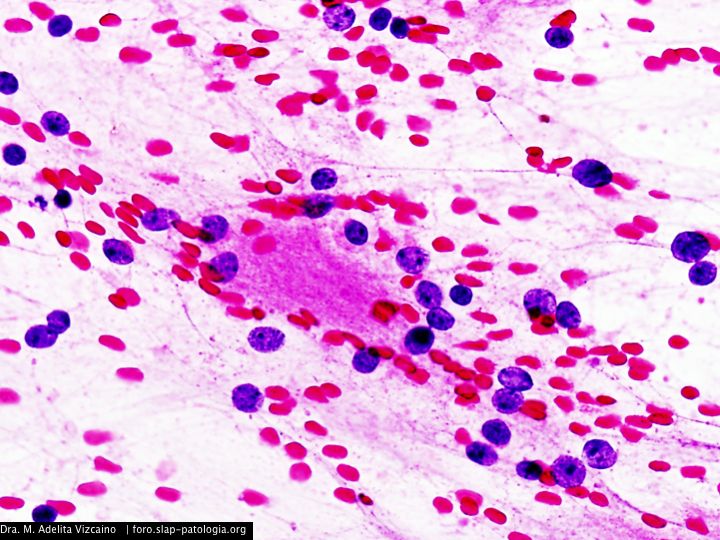
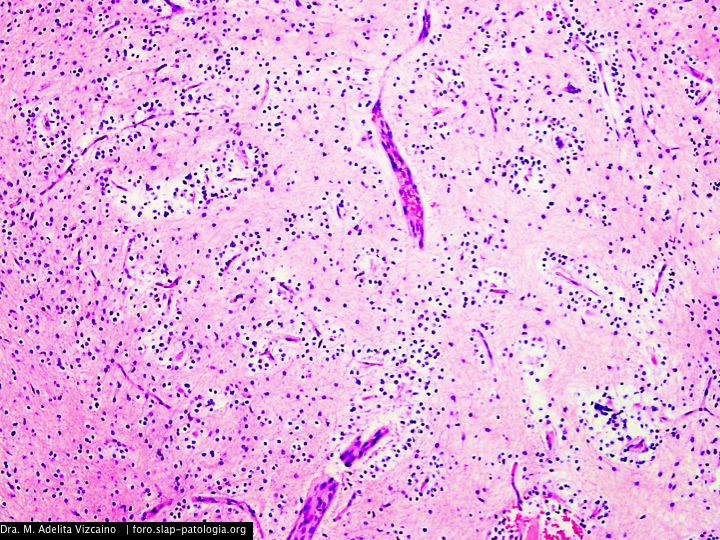
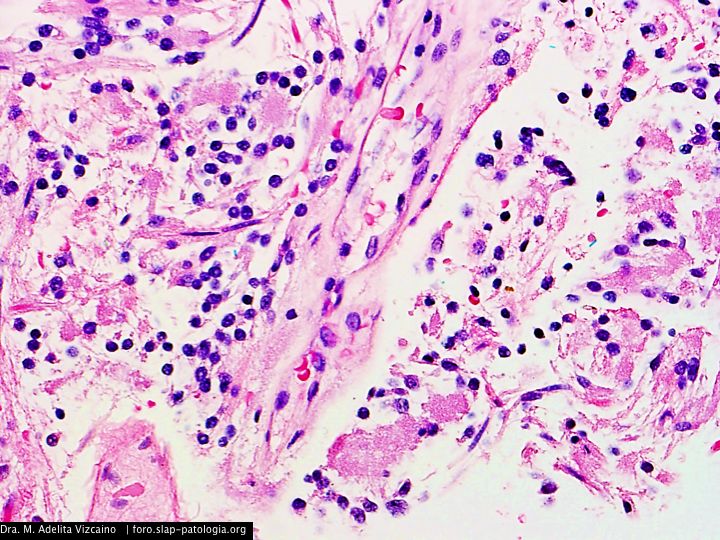

Cual es su diagnostico?
DIAGNOSTICO:
Tumor glioneuronal formador de rosetas.
Patología
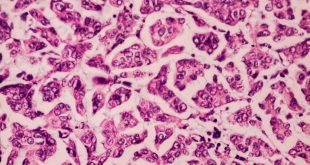
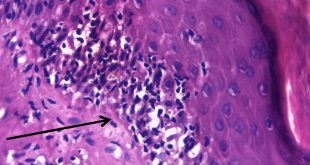
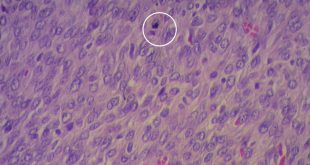
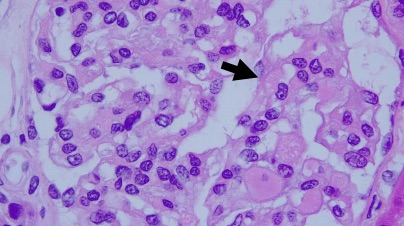
El tumor, localizado en el cuarto ventrículo, fue extirpado y analizado histológicamente. Se caracteriza por una proliferación hipocelular constituida por células que tienen núcleos esféricos, sin pleomorfismo, que alternan con otras que poseen núcleos ovales y procesos citoplásmicos gliales. El hallazgo distintivo es la presencia de rosetas compuestas por neurópilo central y discretamente eosinófilo, rodeado por células con apariencia neurocítica, características presentes tanto en las improntas como en el material fijado. El índice de proliferación con Ki-67 es muy bajo (<1%). Discusión
El tumor glioneuronal formador de rosetas es una neoplasia infrecuente grado I de la Organización Mundial de la Salud (OMS) descrita por Komori y colaboradores en 2002 [1], cuya incidencia se desconoce debido al reducido número de casos reportados hasta la fecha. Su edad de presentación promedio es de 31 años, con ligero predominio en mujeres. Típicamente se presenta como una lesión de crecimiento lento localizada en la línea media de la pared del cuarto ventrículo, que hace prominencia hacia la luz ventricular y se extiende hacia el vermis cerebeloso, acueducto cerebral y/o tronco encefálico, por lo que generalmente se manifiesta con datos de hipertensión intracraneana, cefalalgia y ataxia lentamente progresivas. Sin embargo, se han informado casos originados en puente, glándula pineal, tálamo, nervio óptico, entre otros. El pronóstico es excelente y puede ser curado con resección total.
Histológicamente, es una lesión bifásica constituida por células con diferenciación glial (inmunorreactivas para PFAG, OLIG2) que alternan con otras que tienen diferenciación neurocítica (positivas para sinaptofisina, particularmente en las rosetas) [1]. El componente glial es habitualmente el que predomina y tiene características morfológicas similares al astrocitoma pilocítico, en el que los microquistes, fibras de Rosenthal, cuerpos granulares eosinofílicos y calcificaciones pueden ser evidentes. Por su parte, en el componente neurocítico se distingue la formación de rosetas, algunas perivasculares. La necrosis y actividad mitósica suelen estar ausentes, con índices de proliferación (Ki-67) bajos.
El diagnóstico diferencial del tumor glioneuronal formador de rosetas incluye tumores neuronales y glioneuronales (tumor glioneuronal papilar, liponeurocitoma cerebeloso y “tumor glioneuronal con islas similares a neurópilo [rosetadas]”) y gliomas de bajo grado. El tumor glioneuronal papilar tiene histología y fenotipo particulares, con células gliales que forman las papilas y un componente neurocítico entre ellas, además de tener predilección por los hemisferios cerebrales, en comparación con el tumor glioneuronal formador de rosetas. El “tumor glioneuronal con islas similares a neurópilo (rosetadas)” es en realidad un patrón histológico asociado con gliomas difusos convencionales, y frecuentemente presentan mutaciones en IDH1. Con respecto a las neoplasias gliales, la identificación de las rosetas características distinguen al tumor glioneuronal formador de rosetas del astrocitoma pilocítico, ependimoma y oligodendroglioma.
Con respecto a la patología molecular, el tumor glioneuronal formador de rosetas es distinto de otros tumores. Las alteraciones de BRAF e IDH1, características del astrocitoma pilocítico y gliomas difusos, respectivamente, están generalmente ausentes. Por otro lado, recientemente se han detectado mutaciones en los oncogenes FGFR1 y PIK3CA en esta neoplasia [3,4].
References
1-Komori T, Scheithauer BW, Hirose T. A rosette-forming glioneuronal tumor of the fourth ventricle: infratentorial form of dysembryoplastic neuroepithelial tumor? Am J Surg Pathol. 2002;26:582-91.
2-Schlamann A, von Bueren AO, Hagel C, Zwiener I, Seidel C, Kortmann RD, Müller
K. An individual patient data meta-analysis on characteristics and outcome of patients with papillary glioneuronal tumor, rosette glioneuronal tumor with neuropil-like islands and rosette forming glioneuronal tumor of the fourth ventricle. PLoS One. 2014;9(7):e101211.
3-Gessi M, Moneim YA, Hammes J, Goschzik T, Scholz M, Denkhaus D, Waha A, Pietsch T. FGFR1 mutations in Rosette-forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol. 2014;73:580-4.
4-Ellezam B, Theeler BJ, Luthra R, Adesina AM, Aldape KD, Gilbert MR. Recurrent PIK3CA mutations in rosette-forming glioneuronal tumor. Acta Neuropathol. 2012;123:285-7.

En mi opinión con actividad proliferativa ausente, puede tratarse de un gangliocitoma displasico tipo hamartoma.